Nonvesicular GABA release due to reversal of the GABA transporter
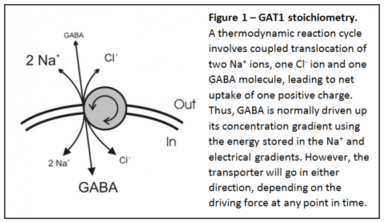
GABA is the major inhibitory neurotransmitter in the brain. We study how GABA is released from neurons and glia, and how this release is affected by anticonvulsants. In addition to classical vesicular release from neurons due to fusion of synaptic vesicles, many neuro-transmitters, including GABA, can also be released from neurons and glia when their uptake transporter operates in reverse.
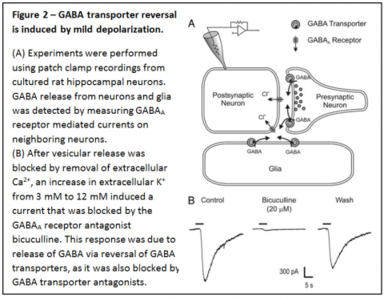
This carrier-mediated GABA release has been recognized to exist for many years, but the physiological relevance has been unclear. Since the GABA transporter is electrogenic and sodium-dependent (Figure 1), this nonvesicular form of GABA release can be induced by high intracellular sodium and depolarization. We have shown that GABA transporter reversal occurs surprisingly easily - in response to mild depolarization induced by an increase in extracellular K+ to as little as 6-12 mM (Figure 2).
Relevance to seizures
There is normally an increase in extracellular K+ during a seizure. Along with the increase in intracellular Na+ and depolarization that also occur during seizures, it is expected that carrier-mediated GABA release would be greatest during seizures, and this would act to reduce excitability. The anticonvulsant vigabatrin inhibits GABA transaminase, the enzyme that metabolizes GABA. This leads to an increase in cytosolic [GABA], which would favor reversal of the GABA transporter. Indeed, we found that vigabatrin induces nonvesicular GABA release, which would inhibit seizures.
Role of the GABA transporter in tonic inhibition
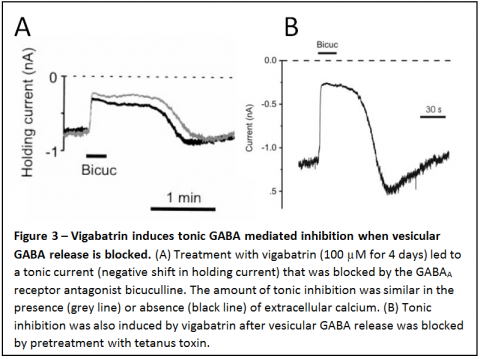
In addition to the effect of vigabatrin in enhancing transporter reversal, this drug also induces a large tonic, GABA mediated inhibition of neurons (Fig. 3). This effect of vigabatrin is very potent, with as little as 50 nM vigabatrin inducing significant tonic inhibition. It is also slow to develop (peaking after 3-4 days), consistent with slow accumulation of GABA in the cytosol. We have shown that tonic inhibition can be due to continuous, spontaneous GABA transporter reversal under some conditions. However, more importantly than the fact that it can occur due to transporter reversal is the following.
![Figure 4 - Extracellular [GABA] at equilibrium increases in response to an increase in intracellular [GABA]. Extracellular [GABA] will increase in response to depolarization as well as in intracellular [GABA] (e.g. after treatment with vigabatrin). At resting potential, extracellular [GABA] would be high enough in some cells at baseline to activate high affinity extrasynaptic GABAA receptors. An increase in cytosolic [GABA] would increase the extracellular [GABA] at equilibrium, and thus tonic inhibition.](/sites/richerson.lab.uiowa.edu/files/styles/no_crop__384w/public/2024-02/4.png?itok=KH7ZAOY-)
An increase in cytosolic [GABA] would lead to a shift in the equilibrium for the transmembrane GABA gradient to a higher extracellular [GABA], as shown in Figure 4. With all else being equal, an increase in intracellular [GABA] would lead to a proportional increase in extracellular [GABA] at steady state.
If extracellular [GABA] is sufficiently high this would activate high affinity, extrasynaptic GABA receptors, leading to tonic inhibition. This type of tonic inhibition is now well recognized to play an important role in regulation of brain excitability. Thus, the increase in tonic inhibition induced by vigabatrin may well be the major reason why this drug prevents seizures.
Neurotransmission can occur due to reversal of the GABA transporter
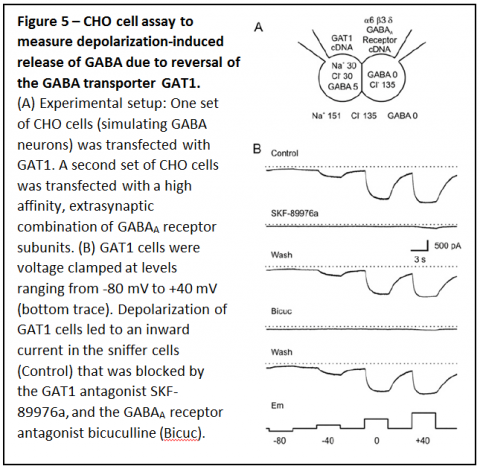
To measure the influence of ambient conditions on transport of GABA by GAT1 we performed experiments in which one set of Chinese Hamster Ovary (CHO) cells was transfected with GAT1, the neuronal form of the GABA transporter (Figure 5A). A separate set of CHO cells was transfected with the a6, b3 and d subunits of the GABAA receptor, a high affinity combination found in some extrasynaptic sites. The GAT1 expressing CHO cells simulated GABAergic neurons, and contained physiological levels of cytosolic sodium and GABA, and the CHO cells expressing GABA receptors were used as “sniffer cells” to detect GABA release. These experiments demonstrated that depolarization over a physiological range caused reversal of GAT1 and the GABA release that occurred induced a substantial response in the sniffer
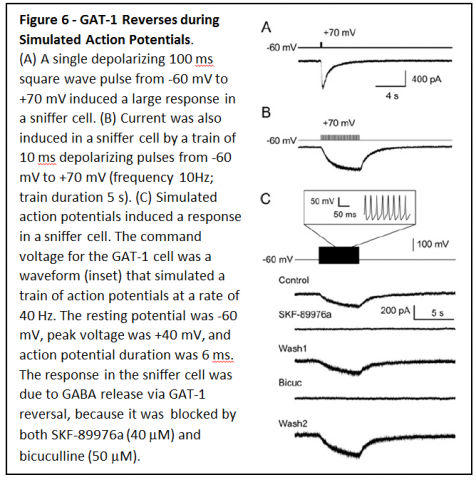
cells (Figure 5B). To determine whether GAT1 can reverse fast enough for GABA release to occur during an action potential, membrane potential of CHO cells expressing GAT1 cells was changed rapidly (Figure 6A & B). Pulses lasting only 10 ms resulted in substantial GABA release that was enough to induce large responses in sniffer cells. When GAT1 expressing CHO cells were depolarized with a waveform that simulated neuronal action potentials (Figure 6C), GAT1 reversal was rapid enough that it led to large amounts of GABA release.
Based on these results, it is predicted that GAT1 reversal should occur during action potentials in GABAergic neurons, and should allow nonvesicular neurotransmission between neurons. To determine whether that occurs, patch clamp recordings were made from hippocampal neurons in primary cell culture.
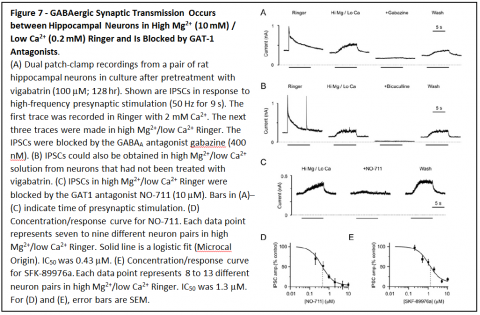
Recordings were made simultaneously from presynaptic GABAergic neurons and postsynaptic neurons. Stimulation of action potentials in presynaptic neurons led to responses in postsynaptic neurons that were blocked by GABAA receptor antagonists bicuculline and gabazine, and by GAT1 antagonists NO-711 and SKF-89976a. Similar results were also obtained when vesicle fusion was blocked by tetanus toxin and when neurotransmitter loading of vesicles was blocked by concanamycin A.
See complete results in the following publications:
Wu, Y., W. Wang, A. Diez-Sampedro & G.B. Richerson. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56(5): 851-65, 2007.
Richerson, G.B. & Y. Wu. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol 90:1363-1374, 2003. [PMID: 12966170]
Wu, Y., W. Wang & G.B. Richerson. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J Neurophysiol 89(4): 2021-2034, 2003.
Wu, Y., W. Wang & G.B. Richerson. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. J Neurosci 21(8):2630-2639, 2001.
Gaspary, H., W. Wang, & G.B. Richerson. Carrier-mediated GABA release induces GABA receptor conductances in hippocampal neurons. J Neurophysiol 80:270-281, 1998.